
The U.S. Food and Drug Administration (FDA) has two major objectives: (1) “assuring the safety, efficacy, and security of human and veterinary drugs” and other medical interventions, and (2) “advancing public health by helping to speed product innovation.” These aims are addressed through research and clinical trials, a sometimes lengthy process designed to clearly assess the safety and efficacy of experimental interventions. However, these two charges can conflict at times as the public pressures the FDA for quick access to new treatments and interventions yet also expect approved treatments to be safe.
A major movement of patient advocates has worked since 2014 to hasten access to experimental interventions in the U.S. As a result, a number of state legislatures have passed “Right to Try” laws, which are designed to give terminally ill patients access to early investigational drugs before full FDA approval and before they are available under the FDA’s “expanded access” policy. Right to Try laws highlight a growing conflict between patients and the FDA, and arguably make the safety and efficacy of unproven drugs secondary to speedy access during the test phase.
In this paper, we review the background and purpose of the FDA, the clinical trials process, and the history of the FDA’s expanded access policy as well as the Right to Try movement and the collection of laws it has produced. Finally, we discuss how Right to Try laws impact the FDA and patients’ rights in the United States and recommend ways to promote faster access to safe treatments.
Promoting Safe and Effective Drugs and Therapies: The Emergence of the FDA
The FDA’s origins date to the turn of the 20th century. Initially called the Division of Chemistry at the U.S. Department of Agriculture, the agency was subsequently named the Bureau of Chemistry and became the Food, Drug, and Insecticide Administration in 1927 (FDA 2014; FDA 2009a). In 1930, its name was shortened to the Food and Drug Administration. Since its inception, the FDA’s mission has been reviewed and altered several times, but it has always served as the primary channel through which drugs, medical devices, and biologics1 are validated.
The agency started under the purview of the Federal Food and Drugs Act of 1906, which states that altered or misbranded drugs or foods cannot be sold or distributed in the United States (FDA 2009a). After products marketed as drugs and cosmetics were shown to be dangerous, Congress passed the Federal Food, Drug, and Cosmetic Act (FDCA) in 1938. This law specifically requires that drugs be labeled with the names of compounds it contains and directions for proper use. The FDCA also mandates that new drugs be proven safe before going to market (FDA 2012; FDA 2015b).
The FDA was again a focus of legislation in 1962 when Congress passed the KefauverHarris Drug Amendments in response to the thalidomide tragedy. Although thalidomide was never approved in the United States, it was prescribed to pregnant women to alleviate morning sickness in 46 countries, including Canada, the United Kingdom, and Germany. Despite its effectiveness for that purpose, thalidomide caused severe birth defects. As a result, the Kefauver-Harris Drug Amendments require that any drugs entering the market after human trials be effective as well as safe (Fintel et al. 2009; FDA 2009b; FDA 2016a).
Congress passed additional FDA legislation in 2007 and 2012. The FDA Amendments Act of 2007 increases the agency’s ability to conduct reviews of new drugs, including post-marketing studies, safety label changes, and risk assessment (FDA 2011). Five years later, the FDA Safety and Innovation Act of 2012 (FDASIA) introduced the “breakthrough therapy” classification. This designation allows expedited review and production of a drug when preliminary evidence suggests it will help terminally ill patients. FDASIA also clarifies guidelines for the approval of new biological devices (FDA 2015c).

The FDA’s mission currently includes both the protection of public health and the assurance of rapid innovation (Box 1). However, these two charges can conflict at times. Patients may advocate for drug approval prior to the end of clinical trials, while regulators want to make sure its risks and efficacy are adequately assessed. Early results might hide major risks or side effects, which can only be detected after a larger trial is conducted.
Recent examples of drugs that were approved by the FDA but that ultimately resulted in serious side effects unforeseen in initial trials include Avastin and Vioxx. Avastin was approved to treat metastatic colorectal cancer. Following promising results from one study, it also was conditionally approved for breast cancer through the accelerated drug program (Hamburg 2011; Huzwitz 2004; National Cancer Institute 2014). Unfortunately, subsequent research in 2011 found that Avastin did not provide enough of a benefit to outweigh the risks, which include stroke, congestive heart failure, and kidney failure (NCI 2014). The FDA removed breast cancer from the drug’s label to ensure it is no longer prescribed to treat breast cancer.2 Vioxx, approved in 1999 for osteoarthritis, was marketed as a miracle drug. It was pulled from the market by 2004 after clinicians found the drug significantly increased the risk of heart attack and stroke (FDA 2013a; FDA 2009c). These examples and many others are reminders of the importance of clinical trials and adequate post-market surveillance.
Clinical Trials
A clinical trial is defined as “a prospective study comparing the effect and value of intervention(s) against a control human being” (Friedman et al. 2010). It is the cornerstone of drug and medical device review by the FDA and other national regulatory agencies.
Traditionally, clinical trials progress from Phase 1 to 3; most drug or device tests on humans start at Phase 1. Participants in Phase 1 trials include not only healthy volunteers but also patients who have tried every other standard treatment or who have tried all available treatments for their condition without success. Phase 1 studies focus on the safety of the intervention and the maximum tolerable dose of the drug (Friedman et al. 2010). This phase consists of small group of people; the length of this phase depends on the intervention being tested.
If the Phase 1 trial is successful the drug continues to Phase 2, which focuses on biological activity or the effect on a patient. This phase includes a control group that receives a placebo; in Phase 1 trials, all volunteers receive pre-assigned dosages of the tested drug. Typical Phase 2 trials involving cancer patients have fewer than 30 participants (Friedman et al. 2010).
Phase 3 trials build on the results of Phase 2 studies to determine the effectiveness of the treatment and its use in a clinical setting involving a much larger number of patients— sometimes hundreds to thousands of patients (Richardson 2015). However, the follow-up period in Phase 3 studies is relatively short compared to the prescribed length of actual treatment for a patient.
Phase 4 trials, started in the 1970s, are relatively uncommon and focus on the longterm effectiveness and safety of a drug or device after it has been released to the market (Friedman et al. 2010; Darrow et al. 2014). When organizing a Phase 4 trial, there are unique issues to address that the other phases may not encounter. For instance, Phase 4 trials need to identify metrics that ensure the company is following the impact of the drug in question as opposed to that of other drugs the patient may be using. Such questions may be extremely difficult or impossible to address (Hill 2012).
In 2006 the FDA introduced a new early exploratory approach, or Phase 0 trials. These “exploratory investigational new drug studies,” which involve limited human exposure, look at the effect of small doses of the drug on patients and how they are metabolized. It is especially useful when animal data is not available. These studies are uncommon and are not focused on therapeutic results (U.S. Department of Health and Human Services 2013). The goal is to determine problems with the drug, such as side effects, to prevent the waste of time and money later in Phase 2 or 3 trials (American Cancer Society 2014a).
Patients Advocate for Increased Access to Experimental Drugs
It often takes years to gain FDA approval for a new drug or device. Patients may die waiting for a drug to come to the market if they do not qualify for a trial. This has led patients to seek out experimental drugs on their own, sometimes through illegal means. Additionally, patients have pressured the FDA to accelerate the clinical trial process and to expand access.
The expanded access movement started in the 1970s when terminally ill patients in the U.S. tried to obtain Laetrile, a drug once used worldwide to treat cancer but that was unavailable in the United States (NCI 2015). Patient advocates sued for access to the drug, claiming that terminally ill patients were exempt from FDA regulations. The U.S. Supreme Court rejected their argument and blocked access to the drug pending FDA approval (United States v. Rutherford 1979). In the end, Laetrile was proven to be ineffective and was never approved by the FDA (NCI 2015).
Although the first attempt by patient advocates to gain access to unproven interventions failed, they continued to push. In the 1980s they succeeded, getting early access to AIDS medications. At the time, the drug Azidothymidine (AZT) was approved for AIDS patients, but it had not been tested on HIV-positive patients who had not yet developed AIDS. Clinical trials finally started in 1986, but patients feared placement in the placebo group (Chassion et al. 1986). Because of the limited access, HIV-positive patients started obtaining the drug from “grey” markets where other patients sold their pills or shared the ones they got in trials (Leonard 2009). This impacted the legitimacy of the trial process and ultimately resulted in the creation of the FDA’s “compassionate care” program, later renamed the expanded or early access program. Through this program, patients can be granted access to experimental drugs that are in Phase 2 or 3 trials if there are no alternative therapies and they are ineligible or unable to participate in a clinical trial (FDA 2016b).
A patient and his or her physician can apply to the expanded access program under the “individual,” the “intermediate,” or the “treatment” category. The main difference between the categories is the number of participating patients. The “individual” program is for one patient, while the “intermediate” category is for a collection of a few hundred patients seeking the same intervention for the same disease according to an FDA grouping (21 CFR 312). The “treatment” category is for a large group of hundreds to thousands of patients who want to access the same drug or device (FDA 2013b; Richardson 2015; Darrow et al. 2015).
For the three expanded access categories the following criteria must be met: (1) the patient’s condition is life-threatening, (2) the benefits of the investigational drug or device outweigh the risks, and (3) the administration of the intervention will not interfere with any aspect of the clinical trial (21 CFR 312). The FDA expanded access program does allow patients with immediately life-threatening diseases to apply for access to drugs that have passed Phase 1 trials (Dresser 2015). Even so, the FDA requires data suggesting the drug is promising enough to give to a patient (Dresser 2015).
The FDA estimates that an expanded access program application requires approximately 100 hours to complete; this includes 26 separate types of information and seven attachments (Rubin 2015; Richardson et al. 2015). Advocates argued that this time commitment strongly discouraged many interested patients and their physicians (Richardson 2015). To address these complaints, the FDA in 2015 released draft guidelines to streamline the process. The new FDA application would require only eight pieces of information and one attachment—reducing the time needed to complete the form to only 45 minutes (U.S. DHHS 2015; Rubin 2015). Furthermore, the FDA by statute will review new applications within 30 days of receipt. If the patient may die in the next 30 days, physicians may submit the form and obtain provisional approval via telephone, on the condition the written application is submitted within 15 days (U.S. DHHS 2015).
Before reaching the FDA, the application is evaluated by an institutional review board (IRB). An IRB is an independent committee that examines investigations involving human subjects. In the case of the expanded access program, the IRB reviews research protocols, including assessing the risks and benefits to safeguard patients. The IRB also addresses the language and terminology used in consent forms, and can even waive the requirement of a patient’s signature on a consent form if the harm of the intervention is minimal (Enfield and Truwit 2008; 21 CFR 56).
In addition to FDA approval for expanded access, manufacturers must agree to provide the drug or biological device to the patient. This can be a stumbling block, as the FDA cannot compel companies to provide expanded access, and many companies disfavor sharing the drug/device extensively as it would limit its availability for the clinical trial, which is their higher priority. Obtaining the drug/ device from the manufacturer tends to be the limiting step in the expanded access program (Darrow et al. 2015). Manufacturers who agree to do so may charge a fee or provide the drug/device for free.
Despite the expanded access program at the FDA, patient advocate groups such as the Abigail Alliance are working to gain even better access to unproven drugs for the terminally ill. In 2003, the Alliance submitted a citizens’ petition to the FDA requesting patient access to experimental therapies after Phase 1 testing without FDA involvement. The group’s due process argument was straightforward: terminally ill patients have a constitutional right to access life-saving drugs even if the drugs have not passed all testing phases (Leonard 2009). While the U.S. Court of Appeals for the District of Columbia agreed with the Abigail Alliance, the Supreme Court ruled in favor of the FDA (Leonard 2009; Abigail Alliance for Better Access to Developmental Drugs v. Von Eschenbach 2007).
After such failures in the courts, early access advocates turned to the U.S. Congress for assistance, but were also unsuccessful. For instance, in 2008 Sen. Sam Brownback of Kansas introduced the Access, Compassion, Care, and Ethics for Seriously Ill Patients (ACCESS) Act; under the act, companies could charge patients for early access drugs and medical professionals were protected from liability lawsuits filed by patients or family members (Leonard 2009). In 2014, Rep. Morgan H. Griffith of Virginia introduced the Compassionate Freedom of Choice Act, which would grant terminally ill patients access to investigational drugs without FDA-required paperwork (Richardson 2015). Congress passed neither law. Interestingly, social media campaigns have had some success in pressuring companies like Johnson & Johnson to start new or additional clinical trials that include terminally ill patients requesting treatment (Caplan and Moch 2014).
The Right to Try Movement
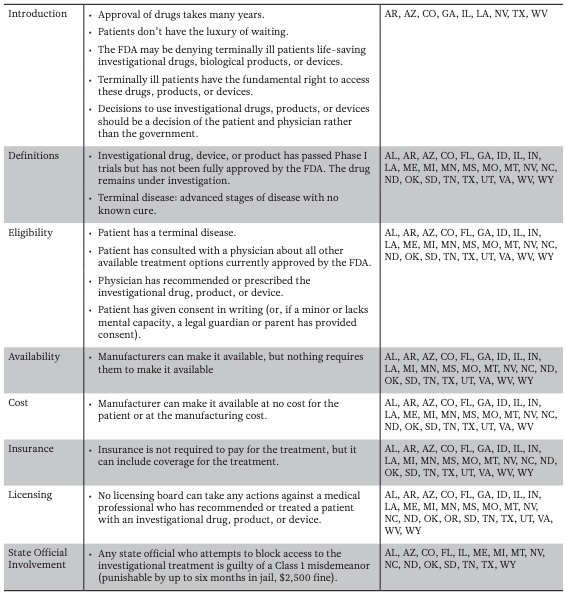
Efforts to increase access to unproven medical interventions have most recently involved state legislatures across the country passing laws affirming a patient’s “right to try.” The Right to Try movement originated at the Goldwater Institute, a conservative and libertarian public policy think tank based in Arizona (Bernick 2016). In 2014, the institute outlined proposed legislation that would give terminally ill patients the “right to try” investigational drugs/ devices still in the testing phase, without FDA involvement. (Corieri 2014). Patients could get early access if: (1) he or she is terminally ill, (2) his or her physician recommends the drug or device, (3) the patient has provided informed consent, and (4) the drug or device has passed Phase 1 of testing (Corieri 2014; Farber et al. 2015; Table 1a).
Proponents of Right to Try laws say the FDA’s expanded access system takes too much time due to the IRB and the paperwork process. In rural areas, it is particularly difficult to get IRB approval fast enough for terminally ill patients (Corieri 2014). Right to Try laws nullify the requirement for IRB and FDA approval, theoretically removing these roadblocks.
The Goldwater report highlights a “constitutional right to medical autonomy,” an idea that plays a critical role in Right to Try laws. The phrase is from the 1973 U.S. Supreme Court case Doe v. Bolton, which resulted in a ruling that overturned the abortion ban in Georgia. The Goldwater report points to a statement in the ruling declaring that one has the “right to care for one’s health and person.” Although controversial, many states have incorporated aspects of the Right to Try concept into law, asserting that access to investigational drugs is a fundamental right.
States Adopt Right to Try Laws
Colorado became the first state to pass a Right to Try law in May 2014, but in the two years that followed, 27 additional states passed a version of the law—for a total of five in 2014, 19 in 2015, and four in 2016. (Colorado House of Representatives 2014; Servick 2014; Gaffney 2015). California was the first state to reject a Right to Try law. In vetoing the bill in October 2015, Gov. Jerry Brown said that the FDA already allows patients to try experimental drugs. Rather than creating a new system, Brown suggested “[w]e should give this federal expedited process a chance to work” (Siders 2015).
State Right to Try bills are similar to the one proposed by the Goldwater Institute. However, many of the bills include different amendments and modified terminology. Some state-specific amendments include removing hospice care eligibility and limiting financial liability for living family members if the patient dies (Table 1b). The Goldwater description of a late-stage terminal illness—an “advanced stage of an illness with no cure” as determined by the physician—was changed in Illinois to “a disease that, without life-sustaining measures, can reasonably be expected to result in death in 24 months or less” (Illinois House of Representatives 2014). Other states like Florida, Mississippi, and Missouri have included clauses requiring that patients be told if future clinical trials prove the investigational drug/device is ineffective. In Mississippi and Missouri, the drug can no longer be provided if found to be ineffective, but Florida allows the continued use of the drug/device for a different condition (Mississippi Senate 2014; Missouri House of Representatives 2014; Florida House of Representatives 2015).
Table 1 — State Implementation of Right to Try Laws
As of May 2016, 28 states have passed Right to Try laws using the Goldwater Institute proposed law as a model. Most used exact language proposed by Goldwater (Table 1a) but a few states included additional language and amendments (Table 1b).
1A — Goldwater Institute Proposed Right to Try Law
1B — State Specific Amendments to the Goldwater Proposed Right to Try Law
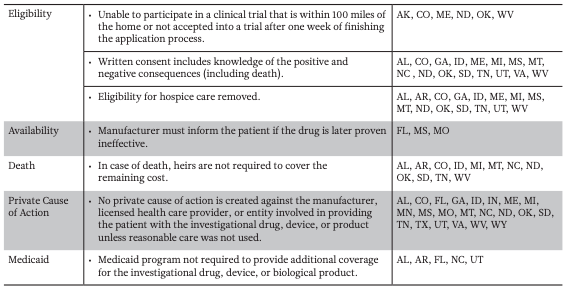
Note Alabama (AL), Arkansas (AR), Arizona (AZ), Colorado (CO), Florida (FL), Georgia (GA), Idaho (ID), Illinois (IL), Indiana (IN), Louisiana (LA), Maine (ME), Michigan (MI), Minnesota (MN), Mississippi (MS), Missouri (MO), Montana (MT), Nevada (NV), North Carolina (NC), North Dakota (ND), Oklahoma (OK), Oregon (OR), South Dakota (SD), Tennessee (TN), Texas (TX), Utah (UT), Virginia (VA), West Virginia (WV) and Wyoming (WY).
Unintended Consequences of Right to Try Laws
The public’s perception of Right to Try laws is mixed. The chance to access investigational drugs can naturally raise the hopes of patients and families. (Turkewitz 2015). However, many policy scholars, physicians, and scientists believe this can be a false hope, and criticize the laws for, at times, causing more stress in a terminally ill patient’s life. Critics also observe that the wording of the Goldwater Institute report and state Right to Try laws focuses more on protecting the physician than ensuring drug/device access to terminally ill patients.
Right to Try laws can perpetuate the idea that an experimental drug is worth the risk and potential danger, despite the fact that 85% of experimental drugs fail during clinical trials (Yang et al. 2015; Ledford 2011). Nonetheless, 76% of Americans think that clinical trials have great value and 73% of Americans are very likely or somewhat likely to participate in clinical trials (Research!America 2013).
Another problem is that Right to Try laws make assumptions about patient and physician knowledge of ongoing clinical trials. The laws assume that patients and physicians can adequately identify experimental drugs that will help them and assess the risk-benefit threshold themselves. Since clinical data is often proprietary and unavailable to physicians, patients and physicians might not have the necessary information to make a fully informed decision of which drug to use (Darrow et al. 2014). It is possible that access to the investigational drug or device will make the patient’s condition worse and exacerbate suffering.
In contrast, the FDA receives updates after each phase of the clinical trial and has information most physicians do not (Friedman et al. 2010).
The laws can also create an expectation of access when, in fact, obtaining the drugs from the pharmaceutical companies is often more difficult than getting expanded access approved by the FDA. Right to Try laws do not ensure that manufacturers will provide the drug or that insurance companies will cover the cost. They effectively give patients the right to beg companies for a drug (Kearns and Caplan 2015). Although critics of the expanded access program say the FDA’s administrative process is the roadblock, the agency approves more than 99% of expanded access requests (U.S. DHHS 2013). Of the 5,849 applications the FDA received between 2010 and 2014, only 33 were denied (Richardson 2015). As noted earlier, the FDA is updating the expanded access application process to reduce the amount of paperwork required, thus the time it takes to complete (US DHHS 2015). Compared to getting FDA approval for expanded access, obtaining experimental drugs from pharmaceutical companies is often the more difficult challenge (Sanghavi 2013; Farber et al. 2015; Neumann 2015).
Those who oppose early access argue that providing an investigational drug/device to a patient who is not part of a clinical trial constrains trial resources and can delay the trial. As a result, the product may not reach the general public as quickly, affecting public health (Dresser 2015). Manufacturers are concerned that if patients receive the drug outside of a clinical trial, enrollment in the trial may drop because patients would rather secure the drug than risk receiving the placebo (Caplan and Bateman-House 2015; Dresser 2015). Moreover, a terminally ill patient with special health circumstances may affect the results of the clinical trial; if weak, terminally ill patients are given the investigational drug, they may suffer more harshly, have a severe side effect, or die. This evidence must be included a side effect of the investigational drug or device. Although the FDA has said that such events would not affect approval, manufacturers are skeptical (Servick 2015; Caplan and Bateman-House 2015).
With the costs of drug development rising to $1 billion to $2 billion per drug, pharmaceutical companies are unlikely to want to gamble with the clinical trial results for an investigational drug or device by allowing access to treatments to patients who do not qualify for the trial (Holbein et al. 2015; Dresser 2015; Tufts Center for the Study of Drug Development 2016).
Several versions of Right to Try laws affect patient benefits, including access to health care and health insurance. The laws passed in Alabama and Colorado, for instance, make patients ineligible for hospice care while undergoing treatment (Table 1b; Alabama House of Representatives 2015; Colorado House of Representatives 2014); once the treatment is over, hospice care can resume. Oklahoma allows insurers to deny coverage during treatment and up to six months after the treatment has ended (Oklahoma House of Representatives 2015). Right to Try laws are intended to enhance the quality of a patient’s life, but these amendments can have a detrimental effect on the patient’s health and finances.
As noted earlier, Right to Try laws do not require manufacturers or insurance companies to pay for the investigational drug or device; the financial burden is placed on patients and their families. This stipulation unintentionally leads to a small, financially privileged group that has access to the drug (Jacob 2015).
An important difference between Right to Try laws and the FDA’s expanded access program is when the drug or device is made available to patients. Right to Try laws permit patients to request drugs that have passed Phase 1 trials, though at that point the drug has not yet been shown to be effective. In contrast, the expanded access program usually provides drugs that have passed Phase 2 or 3 testing, when their effect on patients is better understood. In emergency situations, the FDA will provide access to a drug prior to the end of Phase 1 testing, but this process depends on compliance from the manufacturer (U.S. DHHS 2013; Dresser 2015). Some Right to Try laws require the physician to describe the consequences of taking the drug—good and bad, including death—prior to seeking the intervention. However, physicians may be unable to offer an accurate assessment before the drug passes Phase 2 or 3 clinical trials, leaving the patient potentially uninformed while consenting to receive the intervention (Bateman-House et al. 2015; Holbein et al. 2015). Additionally, without the involvement of the IRB or FDA, no entity exists to verify that the consent form is accurate or comprehensive (Bateman-House et al. 2015).
Right to Try laws focus on terminally ill patients, excluding patients that face debilitating but not immediately life-threatening illnesses such as multiple sclerosis (Bateman-House et al. 2015; Caplan and Bateman-House 2015). The number of people that are excluded from these laws is unknown but could be large (Caplan and Bateman-House 2015).
To help manage the growing number of patients requesting access to drugs in clinical trials, pharmaceutical companies are creating semi-professional panels to decide who receives the drugs outside of the trial. This panel is reminiscent of a Life or Death Committee, informally known as a “God panel.” For instance, in 1961 at the Seattle Swedish Hospital, the God panel decided who would receive a new life-saving treatment, the Scribner shunt for kidney disease, as part of a two-year trial. The committee consisted of a lawyer, a banker, a housewife, and a surgeon (Alexander 1962). Although the chosen patients benefited from the treatment, the ethical dilemma in selecting those who effectively would live thanks to the intervention or die without the intervention put the committee in the awkward position of “playing God.”
In 2015, Johnson & Johnson announced the formation of an institutional bioethics panel, headed by bioethicist Arthur L. Caplan. Like the God panel of the 1960s, this panel decides whether patient requests for investigational drugs or devices should be granted (Maron 2015; Thomas 2015). While there is no guarantee that a patient will be “cured” after receiving the drug, the panel will likely face similar criticisms and confront difficult and uncomfortable decisions.
Policy Recommendations
Although Right to Try laws are intended to provide access to investigational drugs for terminally ill patients, they lack the power to compel pharmaceutical companies to provide the investigational drugs. The goal of the laws is to remove the FDA and ethical oversight required in the expanded access application process. However, these are important oversight mechanisms that can prevent harm and promote informed decision-making. Instead of focusing on removing regulation, patients and advocates should work with the FDA to improve access issues to make the program more effective. The FDA has already started the process of revising its application process to make it less cumbersome.
The FDA has not officially commented on Right to Try laws. However, FDA regulations may take precedence over Right to Try laws under the supremacy clause of the U.S. Constitution. This clause invalidates state laws that contradict federal laws. Even so, if the state law grants more rights or protections than the federal law, the state law will often prevail. In the case of Right To Try laws, however, the state statutes do not in practice provide patients with any more access to drugs that have passed Phase 1 trials than the FDA expanded access program. To truly improve access, amending the federal policy and gaining the support of pharmaceutical companies may have a greater impact.
The presumption that access to drugs after Phase 1 may help terminally ill patients is dangerous. Phase 1 trials provide little information about the actual effectiveness of the drug. Contrary to the desired outcome of expanded access, terminally ill patients may instead suffer more and die sooner. Regardless, patients, physicians, and patient advocates continue to fight for early access. In these situations, instead of working to bypass the FDA, patients, physicians, and regulators should work with the pharmaceutical companies to find ways to improve access. The biggest hindrance to obtaining experimental drugs is often getting the drug from the manufacturer. Efforts should be focused on encouraging companies to increase access. Congress and the FDA can incentivize manufacturers’ participation in expanded access by offering tax breaks if the company participates.
Right to Try laws are written with good intentions, but do not provide patients with a new mechanism to access investigational drugs. Instead, the laws can be harmful to both patients and public good by delaying the testing process. Patient advocates should work with the FDA and pharmaceutical companies to improve the current federal system. This includes incentivizing the companies to provide the drug at little to no cost through federal subsidies or tax breaks for the company during production.
Endnotes
1. Biologics consist of sugars, proteins, or nucleotides. Examples of biologics include vaccines, gene therapy, and allergenics (FDA 2015a).
2. Avastin is still used to treat colorectal cancer because the benefits of the drug are considered greater than the risks.
This material may be quoted or reproduced without prior permission, provided appropriate credit is given to the author and Rice University’s Baker Institute for Public Policy. The views expressed herein are those of the individual author(s), and do not necessarily represent the views of Rice University’s Baker Institute for Public Policy.


